Part 1: Getting Started
Note
At the end of this part of the tutorial, you should have a basic understanding of how to perform different OAK operations on the command line. No programming knowledge is necessary for this part, but some basic understanding of the command line and how to install Python is assumed.
First we will install the package and then we will try some command line examples querying the Drosophila (fruit fly) anatomy ontology (fbbt).
Installation
Install from PyPI
First, create a directory:
cd ~
mkdir oak-tutorial
cd oak-tutorial
Then create a virtual environment:
python3 -m venv venv
source venv/bin/activate
export PYTHONPATH=.:$PYTHONPATH
Note
Make sure you have Python 3.9 or higher
Then install:
pip install oaklib
Run the OAK command
After successful installation, try invoking OAK via runoak:
runoak --help
You should see a list of all commands that are supported by OAK.
You can get help on a specific command:
runoak search --help
See also the Command Line section of this documentation
Basics
Query the OBO Library
Next try using the info command to search for a term in the Drosophila (fruit fly) anatomy ontology (fbbt).
The base command takes an --input (or just -i) option that specifies the input
selector. We will return to the full syntax later,
but one pattern that is useful to know is sqlite:obo:<ontology-id>, which uses the SQL Database Adapter to fetch
and use the SQLite version of an ontology.
To do a basic lookup, using either a name or ID:
runoak --input sqlite:obo:fbbt info 'wing vein'
The first time you run this there will be a lag as the file is downloaded, but after that it will be cached. The results should be:
FBbt:00004751 ! wing vein
You get the same results by specifying the CURIE:
runoak --input sqlite:obo:fbbt info FBbt:00004751
Search
You can use the search command to search for terms. You can also use a special search syntax like this:
runoak -i sqlite:obo:fbbt search 't^wing vein'
Note
We switched from --input to the shorter -i form. We will continue to use the abbreviation in this tutorial.
It is up to you which one you use. Some people prefer more verbose explicit options (and the extra typing!). Others
prefer the more compact form. For the whole command line interface we attempt to follow common standards to avoid
any surprises.
Here t means “term” (search in all term fields) and ^ means “starts with” (don’t worry if this sounds a
bit abstract just now, this will be introduced in more detail later).
This will give results like:
FBbt:00004751 ! wing vein
FBbt:00004754 ! axillary vein
FBbt:00004759 ! wing vein L1
FBbt:00004760 ! wing vein L2
...
Note that “axillary vein” matches because this term has an alias
If you want to instead find any terms that contain the string “wing vein”,
then you can use the ~ symbol:
runoak -i sqlite:obo:fbbt search 't~wing vein'
The results should include the previous results, and include broader matches such as:
...
FBbt:00046009 ! presumptive wing vein L1
FBbt:00046030 ! presumptive wing vein L2
FBbt:00046031 ! presumptive wing vein L3
...
You can use the / symbol to perform a regular expression search:
runoak -i sqlite:obo:fbbt search 't/^wing vein L\d+$'
FBbt:00004754 ! axillary vein
FBbt:00004759 ! wing vein L1
FBbt:00004760 ! wing vein L2
FBbt:00004761 ! wing vein L3
FBbt:00004762 ! wing vein L4
FBbt:00004763 ! wing vein L5
FBbt:00004764 ! wing vein L6
Working with local files
To work with a local ontology file, you can provide the filename as input:
wget http://purl.obolibrary.org/obo/fbbt.obo
This will create a file fbbt.obo in your directory. This is an OBO Format file that
can be passed in directly:
runoak --input fbbt.obo search 'wing vein'
This should give the same results as when you used the sqlite adapter.
Introduction to graphs and trees
Fetching ancestors
Next we will try a different command, plugging in an ID (CURIE) we got from the previous search.
We will use the ancestors command to find all subclass-of (rdfs:subClassOf) and part-of (BFO:0000050) Ancestors of ‘wing vein’.
runoak --input sqlite:obo:fbbt ancestors FBbt:00004751 --predicates i,p
You should see body parts such as cuticle, wing, etc, alongside their ID:
...
FBbt:00004729 wing
FBbt:00007000 appendage
...
Predicate Abbreviations
Here we are providing the Predicates to traverse via the -p/--predicates argument.
The values i and p for the predicates argument are short-hand names for
rdfs:subClassOf and BFO:0000050, respectively.
You can get the same effect with the full predicate CURIEs, rdfs:subClassOf and BFO:0000050.
runoak --input obolibrary:fbbt.obo ancestors FBbt:00004751 --predicates rdfs:subClassOf,BFO:0000050
Possible short-hand names are:
- i for the rdfs:subClassOf predicate
- p for the BFO:0000050 predicate
- e for the owl:equivalentClass predicate
Ancestor Statistics
In the previous example we saw that wing and appendage are ancestor concepts of wing vein but we don’t
have any indication of distance. The --statistics option can provide this in a table form:
runoak --input sqlite:obo:fbbt ancestors FBbt:00004751 --predicates i,p --statistics
This generates a TSV table that shows all ancestors plus (a) the number of input terms that count this as an ancestor [only meaningful if multiple inputs provided] (b) minimum distance up from input term to ancestor
id |
label |
visits |
distance |
|---|---|---|---|
FBbt:00004751 |
wing vein |
1 |
0 |
FBbt:00007245 |
cuticular specialization |
1 |
1 |
FBbt:00006015 |
wing blade |
1 |
1 |
FBbt:00007010 |
multi-tissue structure |
1 |
2 |
FBbt:00004729 |
wing |
1 |
2 |
FBbt:00007000 |
appendage |
1 |
3 |
FBbt:00004551 |
adult external thorax |
1 |
3 |
Oak Trees
The tree command will generate an ascii tree for a term
runoak -i sqlite:obo:fbbt tree FBbt:00004751 -p i
* [] FBbt:10000000 ! anatomical entity
* [i] FBbt:00007016 ! material anatomical entity
* [i] FBbt:00007001 ! anatomical structure
* [i] FBbt:00007013 ! acellular anatomical structure
* [i] FBbt:00007245 ! cuticular specialization
* [i] **FBbt:00004751 ! wing vein**
For this example, we show only the is-a tree. You can try other predicates, or even leaving the predicate option unbounded. This will generate large tree displays, due to the facts there are multiple paths to root.
Warning
you may be tempted to pass in only the p predicate to see just the partonomy. However, this will likely generate
a truncated tree, since many parts of are not directly asserted, they must be inferred from an is-a parent.
Later on we will see how to better incorporate reasoning, but for now it is recommended that you always include is-a
as a predicate
Advanced Search
Using search terms as parameters
Search terms can be used as input for any OAK command:
runoak -i sqlite:obo:fbbt tree "t/^wing vein L.*$" -p i
This will feed the search results into the tree command:
* [] FBbt:10000000 ! anatomical entity
* [i] FBbt:00007016 ! material anatomical entity
* [i] FBbt:00007001 ! anatomical structure
* [i] FBbt:00007013 ! acellular anatomical structure
* [i] FBbt:00007245 ! cuticular specialization
* [i] FBbt:00004751 ! wing vein
* [i] FBbt:00047212 ! longitudinal vein
* [i] **FBbt:00004754 ! axillary vein**
* [i] **FBbt:00004759 ! wing vein L1**
* [i] **FBbt:00004760 ! wing vein L2**
* [i] **FBbt:00004761 ! wing vein L3**
* [i] **FBbt:00004762 ! wing vein L4**
* [i] **FBbt:00004763 ! wing vein L5**
* [i] **FBbt:00004764 ! wing vein L6**
Note that the direct matches are highlighted with **...**
Chaining Commands
The output of one command can be passed in as input to another. Just specify - as one of the arguments:
runoak -i sqlite:obo:fbbt search "t/^wing vein L.*$" | runoak -i sqlite:obo:fbbt tree -p i -
This will give the same results as the above
Visualization
Later on we will see how we can use the viz command to make images like:
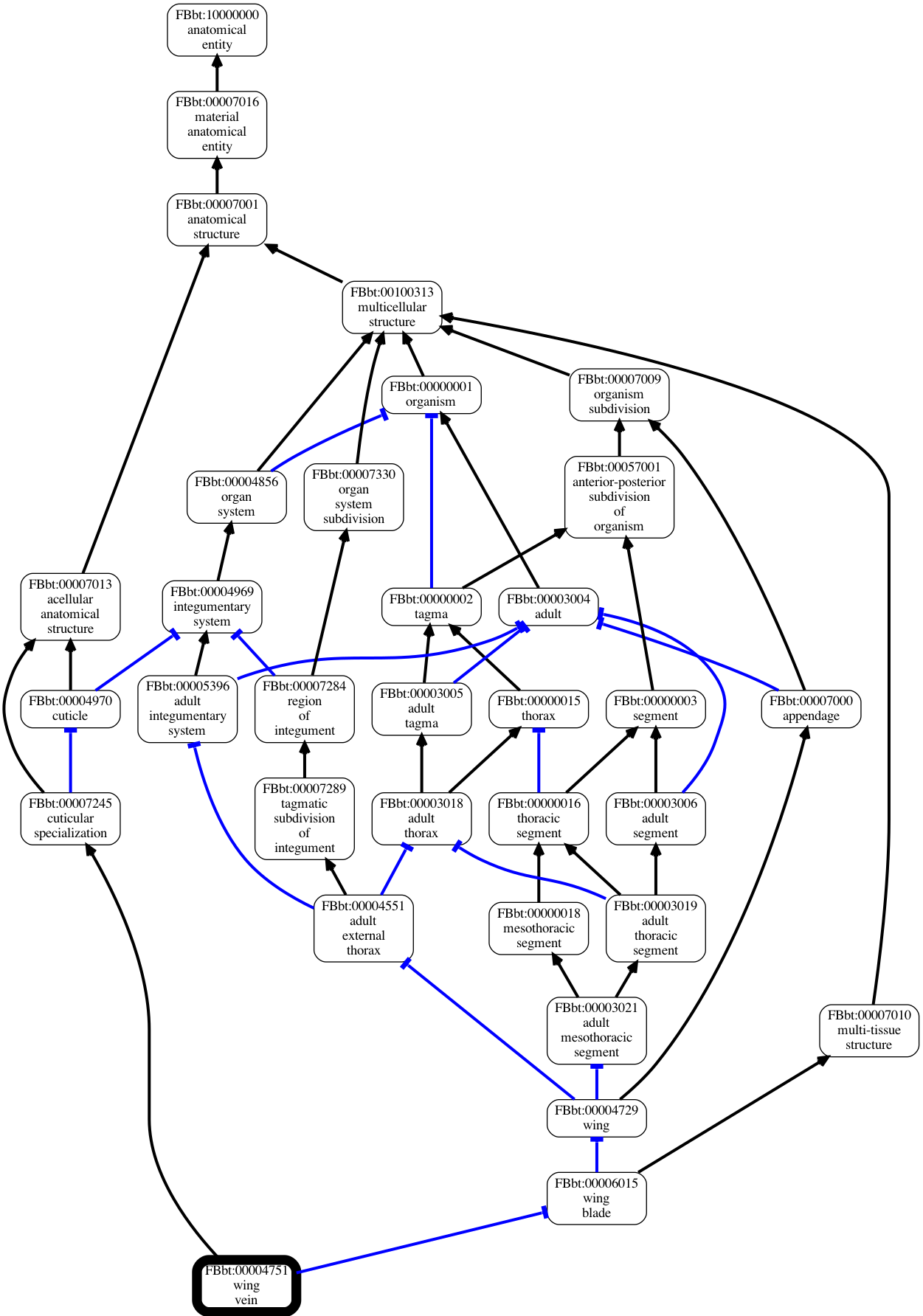
Using other backends
So far we have used SQL Database Adapter.
In fact, OAK allows a number of other backends (also called Implementations). We will give a brief overview of some here
Using Ubergraph
Ubergraph is an integrated ontology store that contains a merged set of mutually referential OBO ontologies.
runoak -i ubergraph: search 'wing vein'
This searches the Ubergraph backend using the blazegraph search interface.
Note that in addition to searching over a wider range of ontologies, this returns a ranked list that might include matches only to “wing” or “vein”. Currently each backend implements search a little differently, but this will be more unified and controllable in the future.
Warning
in future this behavior may change, and relevancy-ranked searching will be more explicitly under control of the user.
You can constrain search to a particular ontology in Ubergraph:
runoak -i ubergraph:fbbt search 'wing vein'
The ubergraph implementation largely allows for the same operations as the SQL one we have seen previously. However, not every implementation implements every operation. And some operations may be more efficient on some implementations. There are a variety of space-time tradeoffs as well. See the Architecture document to learn more.
The main obvious difference is that there is no need for any ontology download - so you can do quick queries:
runoak -i ubergraph:chebi info CHEBI:15356 -O obo
generates obo:
[Term]
id: CHEBI:15356
name: cysteine
def: "A sulfur-containing amino acid that is propanoic acid with an amino group at position 2 and a sulfanyl group at position 3." []
xref: Beilstein:1721406
xref: CAS:3374-22-9
...
is_a: CHEBI:33704 ! alpha-amino acid
is_a: CHEBI:26167 ! polar amino acid
is_a: CHEBI:26834 ! sulfur-containing amino acid
Using Ontobee
Another triplestore you can use is ontobee
runoak -i ontobee:chebi info CHEBI:15356 -O obo
Currently the ontobee implementation does not handle non-isa hierarchical queries.
Using BioPortal and OntoPortal
BioPortal is a comprehensive repository of biomedical ontologies. It is part of the OntoPortal Alliance, which provides access to multiple ontologies outside the life sciences.
To query BioPortal (or any OntoPortal endpoint), first you will need to go to BioPortal and get an API key (if you don’t already have one).
Note
The API Key is assigned to each user upon creating an account on BioPortal.
You will then need to set it:
runoak set-apikey --endpoint bioportal YOUR-API-KEY
This stores it in an OS-dependent folder, which is then accessed by OAK for performing API queries. You don’t need to do this again, unless you switch to a different computer.
After you have set the API key
runoak -i bioportal: search 'wing vein'
Again the results are relevance ranked, and there are a lot of them, as this includes multiple ontologies, you may want to ctrl-C to kill before the end.
Currently the bioportal implementation is not as fully featured as some of the others, and doesn’t take full advantage of all API routes
One of the unique features of bioportal is the comprehensiveness of computed lexical mappings. These can be exported in various SSSOM formats such as yaml or TSV:
runoak -i bioportal:chebi term-mappings CHEBI:15356 -O sssom
The Bioportal endpoint can also be used to Annotate sections of text, for example:
runoak -i bioportal:cl annotate "interneuron of forebrain"
Gives results:
object_id: CL:0000099
object_label: interneuron
object_source: https://data.bioontology.org/ontologies/CL
match_type: PREF
subject_start: 1
subject_end: 11
subject_label: INTERNEURON
---
object_id: UBERON:0001890
object_label: forebrain
object_source: https://data.bioontology.org/ontologies/CL
match_type: PREF
subject_start: 16
subject_end: 24
subject_label: FOREBRAIN
Note that the results here are in YAML syntax, with each result being a YAML document. The results of the annotate command conform to the annotate Datamodel. We will return to the concept of datamodels later on, for now you can look at the Text Annotator Datamodel docs.
Some datamodels can also be expressed as TSVs:
runoak -i bioportal:cl annotate "interneuron of forebrain" -O csv
Gives back a TSV table:
predicate_id |
object_id |
object_label |
object_source |
confidence |
match_string |
is_longest_match |
matches_whole_text |
match_type |
info |
subject_start |
subject_end |
subject_label |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
CL:0000099 |
interneuron |
None |
None |
None |
None |
PREF |
None |
1 |
11 |
INTERNEURON |
||
UBERON:0001890 |
forebrain |
None |
None |
None |
None |
PREF |
None |
16 |
24 |
FOREBRAIN |
Any other implementation that implements the annotate interface will conform to this same datamodel and format.
Using OLS
OLS is a repository of high quality ontologies. It has less breadth than BioPortal. Currently OAK offers very limited functionality with OLS but this will be improved in future.
OLS also aggregates curated mappings, these can be exported in the same way:
runoak -i ols: term-mappings CHEBI:15356 -O sssom
subject_id |
subject_label |
predicate_id |
object_id |
match_type |
subject_source |
object_source |
mapping_provider |
|---|---|---|---|---|---|---|---|
CHEBI:15356 |
cysteine |
skos:closeMatch |
PMID:25181601 |
Unspecified |
CHEBI |
PMID |
CDNO |
CHEBI:15356 |
cysteine |
skos:closeMatch |
PMID:25181601 |
Unspecified |
CHEBI |
PMID |
CHEBI |
CHEBI:15356 |
cysteine |
skos:closeMatch |
CAS:3374-22-9 |
Unspecified |
CHEBI |
CAS |
CHEBI |
CHEBI:15356 |
cysteine |
skos:closeMatch |
PMID:17439666 |
Unspecified |
CHEBI |
PMID |
CHEBI |
CHEBI:15356 |
cysteine |
skos:closeMatch |
KEGG:C00736 |
Unspecified |
CHEBI |
KEGG |
CHEBI |
CHEBI:15356 |
cysteine |
skos:closeMatch |
KNApSAcK:C00007323 |
Unspecified |
CHEBI |
KNApSAcK |
ZP |
CHEBI:15356 |
cysteine |
skos:closeMatch |
Wikipedia:Cysteine |
Unspecified |
CHEBI |
Wikipedia |
ZP |
Next steps
You can play around with some of the other commands (see CLI), or go right into the next section on programmatic usage!