OAK enrichment command
This notebook is intended as a supplement to the main OAK CLI docs.
This notebook provides examples for the enrichment command which produces a summary of ontology classes that are enriched in the associations for an input set of entities.
See also the end of the Command Line Tutorial
Help Option
You can get help on any OAK command using --help
[1]:
!runoak enrichment --help
Usage: runoak enrichment [OPTIONS] [TERMS]...
Run class enrichment analysis.
Given a sample file of identifiers (e.g. gene IDs), plus a set of
associations (e.g. gene to term associations, return the terms that are
over-represented in the sample set.
Example:
runoak -i sqlite:obo:uberon -g gene2anat.txt -G g2t enrichment -U my-
genes.txt -O csv
This runs an enrichment using Uberon on my-genes.txt, using the
gene2anat.txt file as the association file (assuming simple gene-to-term
format). The output is in CSV format.
It is recommended you always provide a background set, including all the
entity identifiers considered in the experiment.
You can specify --filter-redundant to filter out redundant terms. This will
block reporting of any terms that are either subsumed by or subsume a lower
p-value term that is already reported.
For a full example, see:
https://github.com/INCATools/ontology-access-
kit/blob/main/notebooks/Commands/Enrichment.ipynb
Note that it is possible to run "pseudo-enrichments" on term lists only by
passing no associations and using --ontology-only. This creates a fake
association set that is simply reflexive relations between each term and
itself. This can be useful for summarizing term lists, but note that
P-values may not be meaningful.
Options:
-o, --output FILENAME Output file, e.g. obo file
-p, --predicates TEXT A comma-separated list of predicates. This
may be a shorthand (i, p) or CURIE
--autolabel / --no-autolabel If set, results will automatically have
labels assigned [default: autolabel]
-O, --output-type TEXT Desired output type
-o, --output FILENAME Output file, e.g. obo file
--ontology-only / --no-ontology-only
If true, perform a pseudo-enrichment
analysis treating each term as an
association to itself. [default: no-
ontology-only]
--cutoff FLOAT The cutoff for the p-value; any p-values
greater than this are not reported.
[default: 0.05]
-U, --sample-file FILENAME file containing input list of entity IDs
(e.g. gene IDs) [required]
-B, --background-file FILENAME file containing background list of entity
IDs (e.g. gene IDs)
--association-predicates TEXT A comma-separated list of predicates for the
association relation
--filter-redundant / --no-filter-redundant
If true, filter out redundant terms
--help Show this message and exit.
Download example file and setup
We will use the HPO Association file
[2]:
!curl -L -s http://purl.obolibrary.org/obo/hp/hpoa/genes_to_phenotype.txt > input/hpoa_g2p.tsv
next we will set up an hpo alias
[3]:
alias hp runoak -i sqlite:obo:hp
Test this out by querying for associations for a particular gene.
We need to pass in the association file we downloaded, as well as specify the file type (with -G):
[4]:
hp -G hpoa_g2p -g input/hpoa_g2p.tsv associations -Q subject NCBIGene:8192 -O csv | head
subject subject_label predicate object object_label property_values predicate_label negated publications primary_knowledge_source aggregator_knowledge_source subject_closure subject_closure_label object_closure object_closure_label
NCBIGene:8192 None None HP:0001250 None None None None None
NCBIGene:8192 None None HP:0000013 None None None None None
NCBIGene:8192 None None HP:0000007 None None None None None
NCBIGene:8192 None None HP:0010464 None None None None None
NCBIGene:8192 None None HP:0008232 None None None None None
NCBIGene:8192 None None HP:0011969 None None None None None
NCBIGene:8192 None None HP:0004322 None None None None None
NCBIGene:8192 None None HP:0000786 None None None None None
NCBIGene:8192 None None HP:0000815 None None None None None
Enrichment
We will perform enrichment using a set of genes known to be associated with Ehler-Danlos Syndrome (EDS).
The gene list is here:
Let’s take a look at them:
[5]:
!cat input/eds-genes-ncbigene.tsv
id label
NCBIGene:7148 TNXB
NCBIGene:715 C1R
NCBIGene:716 C1S
NCBIGene:126792 B3GALT6
NCBIGene:55033 FKBP14
NCBIGene:91252 SLC39A13
NCBIGene:29940 DSE
NCBIGene:1303 COL12A1
NCBIGene:9509 ADAMTS2
NCBIGene:1278 COL1A2
NCBIGene:1281 COL3A1
NCBIGene:1289 COL5A1
NCBIGene:1290 COL5A2
NCBIGene:84627 ZNF469
NCBIGene:113189 CHST14
NCBIGene:165 AEBP1
NCBIGene:5351 PLOD1
NCBIGene:11285 B4GALT7
NCBIGene:11107 PRDM5
Running the enrichment command
Next we will run the command itself. Note we use two sets of parameters
global OAK parameters:
the format of the associations (
-G), here using the HPOA gene to phenotype formatthe path to the association file (
-g), here the gp2 file we downloaded earlier
local parameters for the
enrichmentcommandthe set of genes to be enriched (via
-Uor--sample-file)the output format for the results (via
-Oor--output-type) - here a TSV, but could also be YAML, RDFthe
--autolabeloption that will do additional HPO queries to give the names of each termthe output file via
-o(--output)
[6]:
hp -G hpoa_g2p -g input/hpoa_g2p.tsv enrichment -U input/eds-genes-ncbigene.tsv -O csv --autolabel -o output/eds-genes-enriched.tsv
Examining the results
The best way to look at TSVs in a notebook such as this one is to use pandas to load as a dataframe. Note however that in most scenarios where you use the command line, this would not be wrapped in a notebook, and you could use your favorite TSV/CSV tool for exploring the results
[7]:
import pandas as pd
[8]:
df=pd.read_csv("output/eds-genes-enriched.tsv", sep="\t")
df
[8]:
| class_id | p_value | class_label | rank | p_value_adjusted | false_discovery_rate | fold_enrichment | probability | sample_count | sample_total | background_count | background_total | ancestor_of_more_informative_result | descendant_of_more_informative_result | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | HP:0000974 | 2.121426e-37 | Hyperextensible skin | 1 | 2.895747e-34 | NaN | NaN | NaN | 19 | 19 | 68 | 5011 | NaN | NaN |
| 1 | HP:0001075 | 1.245666e-36 | Atrophic scars | 2 | 1.700334e-33 | NaN | NaN | NaN | 17 | 19 | 37 | 5011 | NaN | NaN |
| 2 | HP:0008067 | 9.834712e-29 | Abnormally lax or hyperextensible skin | 3 | 1.342438e-25 | NaN | NaN | NaN | 19 | 19 | 177 | 5011 | True | NaN |
| 3 | HP:0100699 | 1.916985e-28 | Scarring | 4 | 2.616685e-25 | NaN | NaN | NaN | 19 | 19 | 183 | 5011 | True | NaN |
| 4 | HP:0004334 | 7.330381e-28 | Dermal atrophy | 5 | 1.000597e-24 | NaN | NaN | NaN | 17 | 19 | 102 | 5011 | True | NaN |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 159 | HP:0010488 | 3.040076e-05 | Aplasia/Hypoplasia of the palmar creases | 160 | 4.149704e-02 | NaN | NaN | NaN | 3 | 19 | 17 | 5011 | NaN | True |
| 160 | HP:0000014 | 3.062457e-05 | Abnormality of the bladder | 161 | 4.180254e-02 | NaN | NaN | NaN | 9 | 19 | 494 | 5011 | True | NaN |
| 161 | HP:0011844 | 3.186952e-05 | Abnormal appendicular skeleton morphology | 162 | 4.350189e-02 | NaN | NaN | NaN | 16 | 19 | 1853 | 5011 | True | NaN |
| 162 | HP:0033353 | 3.286903e-05 | Abnormal blood vessel morphology | 163 | 4.486623e-02 | NaN | NaN | NaN | 12 | 19 | 967 | 5011 | True | True |
| 163 | HP:0025323 | 3.300320e-05 | Abnormal arterial physiology | 164 | 4.504937e-02 | NaN | NaN | NaN | 6 | 19 | 177 | 5011 | True | True |
164 rows × 14 columns
Plotting the results
OAK doesn’t have any command line plotting capabilities yet, but you can easily use external tools. Here we show how to plot the results using matplotlib.
[9]:
import matplotlib.pyplot as plt
import numpy as np
data = df.copy()
# Transform p-values for better visualization
data['negative_log10_p_value'] = -np.log10(data['p_value'])
# Sort data by transformed p-value
data_sorted = data.sort_values('negative_log10_p_value', ascending=True)
# Select top 15 terms with the highest significance
top_terms = data_sorted.nlargest(15, 'negative_log10_p_value')
# Plotting the horizontal bar chart for the top 15 terms
plt.figure(figsize=(10, 10))
plt.barh(top_terms['class_label'], top_terms['negative_log10_p_value'], color='teal')
plt.xlabel('-log10(p-value)')
plt.ylabel('Enriched Terms')
plt.title('Top 15 Enriched Terms by Statistical Significance')
plt.gca().invert_yaxis() # Invert y-axis for better readability
plt.show()
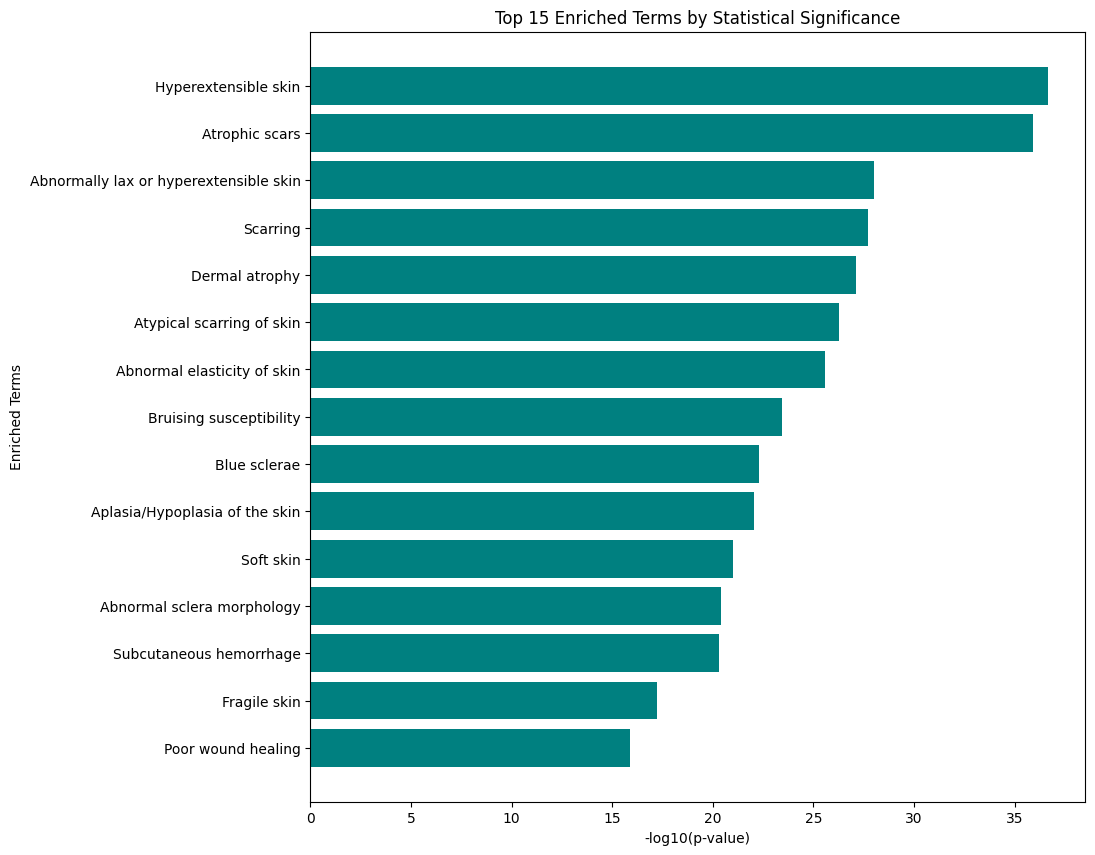
The results here should not be surprising - we already know that hyperextensible skin is a hallmark of EDS, but it’s good to see this confirmed!
We used a somewhat artificial gene set here, but in reality gene sets derived e.g. from gene expression will be more messy and signals weaker.
Visualizing results in a graph form
We can use the OAK viz command to visualize the placement of the enriched terms in the graph.
Because graph visualization can get messy, we’ll take the top 5 results (they are already sorted):
[22]:
!head -5 output/eds-genes-enriched.tsv > output/eds-genes-enriched-top-5.tsv
Next run this through viz. Note that most OAK commands take as input either term lists, or term query lists, where queries can include expressions with functions like .idfile:
[25]:
hp viz -p i .idfile output/eds-genes-enriched-top-5.tsv -O png -o output/eds-genes-enriched-top-5.png
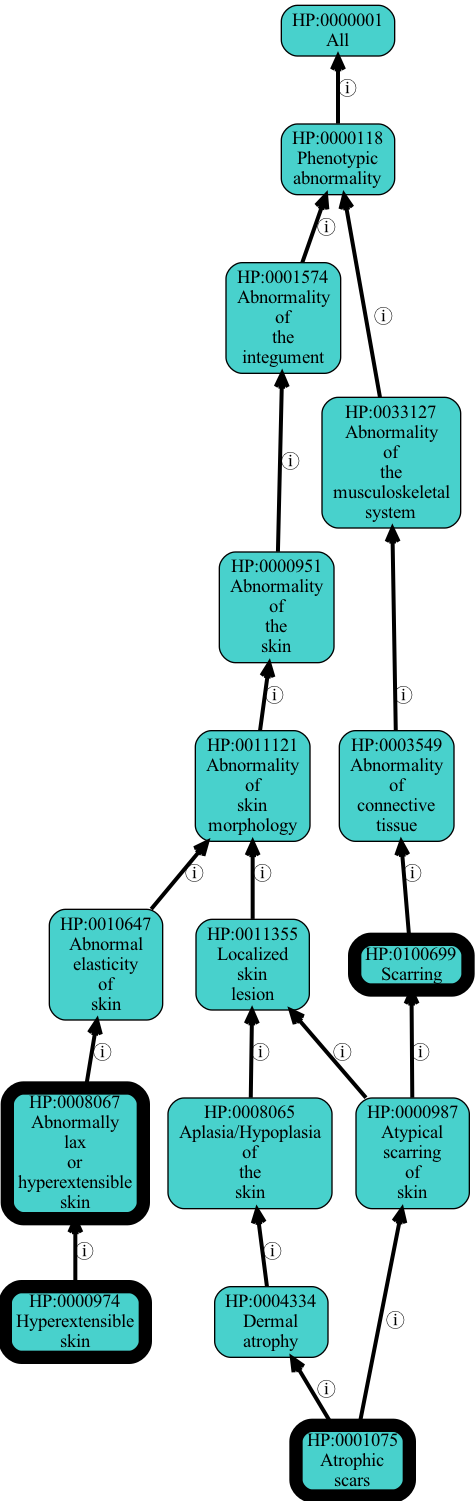
At this time there isn’t an easy way to show the p-value visually, but this may be added later.
When we visualize like this we see there is some redundancy. Scarring is implied by Atrophic scars.
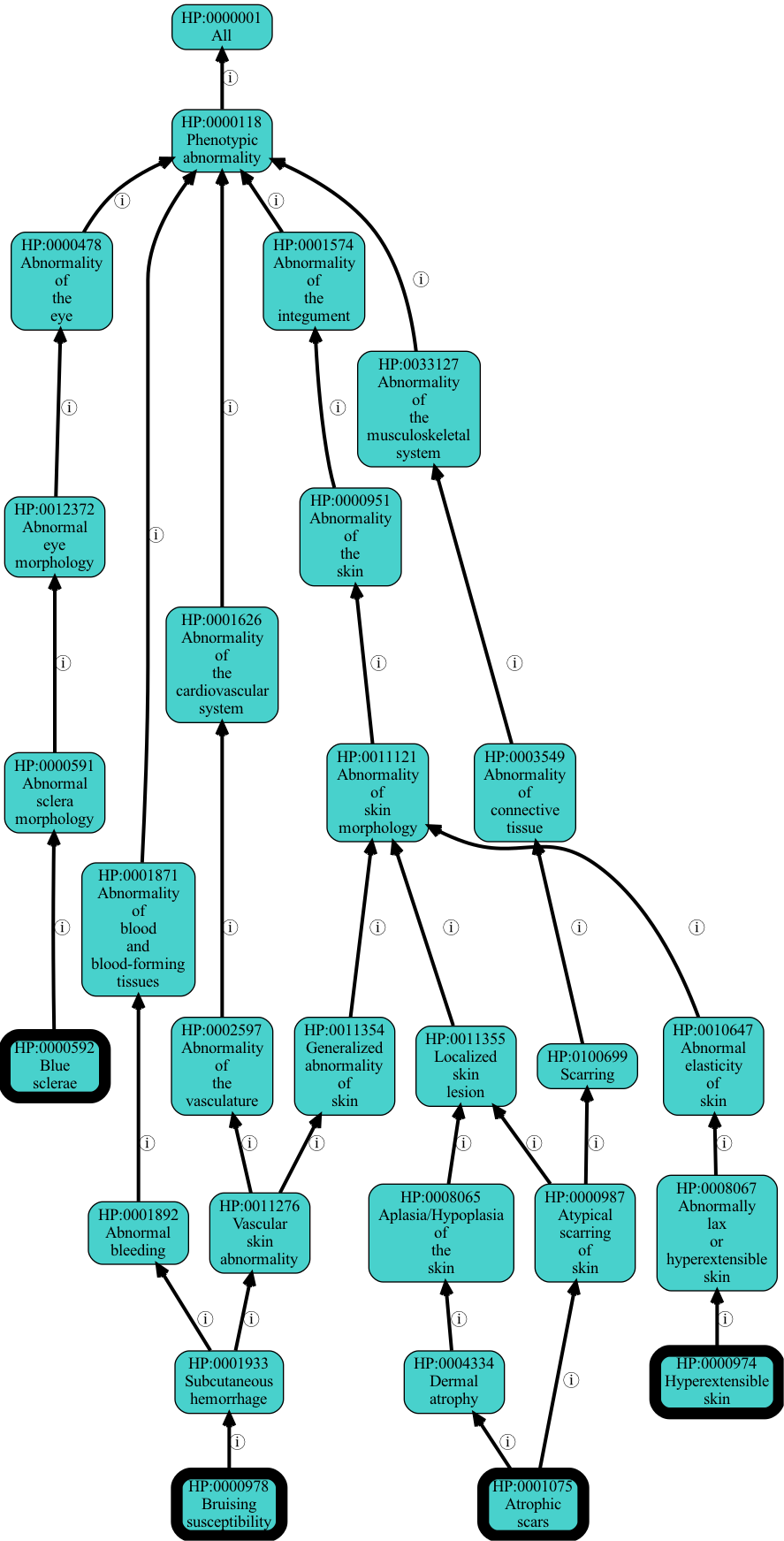
We can run enrichment with --filter-redundant on. This will remove more general terms, unless that term is more significant than all descendants.
[30]:
hp -G hpoa_g2p -g input/hpoa_g2p.tsv enrichment -U input/eds-genes-ncbigene.tsv -O csv --autolabel --filter-redundant -o output/eds-genes-enriched-filtered.tsv
[31]:
!head -5 output/eds-genes-enriched-filtered.tsv > output/eds-genes-enriched-filtered-top-5.tsv
[32]:
hp viz -p i .idfile output/eds-genes-enriched-filtered-top-5.tsv -O png -o output/eds-genes-enriched-filtered-top-5.png
Normalizing input gene lists
What happens if our input gene IDs use a different system than the associations?
E.g. assume our inputs are HGNC and our association file is NCBIGene
Let’s take a look
[11]:
!cat input/eds-genes-hgnc.tsv
id label
HGNC:11976 TNXB
HGNC:1246 C1R
HGNC:1247 C1S
HGNC:17978 B3GALT6
HGNC:18625 FKBP14
HGNC:20859 SLC39A13
HGNC:21144 DSE
HGNC:2188 COL12A1
HGNC:218 ADAMTS2
HGNC:2198 COL1A2
HGNC:2201 COL3A1
HGNC:2209 COL5A1
HGNC:2210 COL5A2
HGNC:23216 ZNF469
HGNC:24464 CHST14
HGNC:303 AEBP1
HGNC:9081 PLOD1
HGNC:930 B4GALT7
HGNC:9349 PRDM5
[19]:
hp -G hpoa_g2p -g input/hpoa_g2p.tsv enrichment -U input/eds-genes-hgnc.tsv -O csv --autolabel
ValueError: No associations found for subjects
As expected, the command complains that no associations were found for any of the subjects (input genes).
So what do we do? There is a separate command normalize that can be used here to normalize an ID list. Normalize can be used with a number of different ID mapper / normalizer sources. Here we are using it with the NCATS Translator SRI node normalizer:
[20]:
!runoak -i translator: normalize .idfile input/eds-genes-hgnc.tsv -M NCBIGene -o output/eds-genes-ncbigene.tsv
[21]:
!cat output/eds-genes-ncbigene.tsv
id label
NCBIGene:7148 TNXB
NCBIGene:715 C1R
NCBIGene:716 C1S
NCBIGene:126792 B3GALT6
NCBIGene:55033 FKBP14
NCBIGene:91252 SLC39A13
NCBIGene:29940 DSE
NCBIGene:1303 COL12A1
NCBIGene:9509 ADAMTS2
NCBIGene:1278 COL1A2
NCBIGene:1281 COL3A1
NCBIGene:1289 COL5A1
NCBIGene:1290 COL5A2
NCBIGene:84627 ZNF469
NCBIGene:113189 CHST14
NCBIGene:165 AEBP1
NCBIGene:5351 PLOD1
NCBIGene:11285 B4GALT7
NCBIGene:11107 PRDM5
This is identical to the gene list in the original example, and can be used in the same way
[ ]: